Understanding USC’s Clinical Trials Office: Support for Research Success
This video introduces USC’s Clinical Trials Office and explains how it supports the successful activation and management of industry-sponsored clinical trials. You’ll learn when to engage CTO, how clinical trials differ from broader clinical research, and why tools and processes like OnCore and Medicare Coverage Analysis are essential for compliance and efficient study startup. By the end, you’ll better understand how to route studies correctly, avoid delays, and support a smoother research process for everyone involved.
How to Register With Clinicaltrials.gov
On September 16th, 2016, The The U.S. Department of Health and Human Services issued a final rule that sets forth expanded requirements for registration and results information to clinicaltrials.gov for FDA-regulated drug, biological, and device products. Simultaneously, NIH issued a complementary policy requiring registration and results information to clinicaltrials.gov for all NIH-sponsored clinical trials, regardless of whether the trial is covered by the HHS Final Rule.
USC’s IRB’s Policy further states:
Registration and results reporting are required for applicable clinical trials; however, ClinicalTrials.gov allows voluntary reporting of other studies that:
- Are in conformance with any applicable human subject or ethics review regulations (or equivalent) and
- Are in conformance with any applicable regulations of the national (or regional) health authority (or equivalent) Investigators may choose to register a study that is not an applicable clinical trial as a condition to publish study results in a journal. FDA regulations require reporting of results from registered trials.
The Responsible Party must generally report results no later than 12 months after the trial completion date. Results must include participant baseline characteristics, participant flow diagram, outcomes, and adverse events. Instructions for submitting results are available at ClinicalTrials.gov. FDA also requires sponsors or investigators to certify compliance with ClinicalTrials.gov registration when submitting certain applications to the FDA. Form FDA 3674 is used to certify compliance.
The text above may be used in NIH applications requiring a statement of USC policy.
Registration is required by law and USC IRB policy for:
- FDA Clinical Trials defined as intervention studies that include drugs, biological products, and medical devices;
- NIH Clinical Trials defined as biomedical and behavioral studies of human subjects;
- Publication of research studies that assigns human subjects to health-related interventions and evaluates their outcomes. Other public registries are also available to meet criteria for ICMJE and WHO; and
- Reimbursement of Medicare claims for items and services in clinical trials that are qualified for coverage.
Registration of an applicable clinical trial must be submitted no later than 21 days after enrollment of the first participant, and the data that must be reported includes
- Participant Flow
- Demographic and Baseline Characteristics
- Primary and Secondary Outcomes
- Results of any Scientifically Appropriate Statistical Tests
- Adverse Event Information
Non-compliance of these requirements may result in civil monetary penalties, withholding/recovery of federal funds, and/or withholding of publications.
Principal Investigators are responsible to register their studies prior to participant enrollment in the Protocol Registration System (PRS).
Contacts to Request a PRS User Account
For Cancer Center Studies
- Tomi Homesy
- Email: Tali.Homsey@med.usc.edu
- Kathya Perez
- Email: Kathya.Perez@med.usc.edu
For Non-Cancer Studies & All Other Requests
- Kimberlee Eudy
- Email: eudy@usc.edu
- Phone: (858) 964-0730
Additional Information
Training Resources
- Final Rule Webinar Series (Overview of the Final Rule & Information Requirements)
- How to Submit Results Data (Overview of Each Results Module)
- Example Studies
STEPS TO REGISTER STUDY ON CLINICALTRIALS.GOV
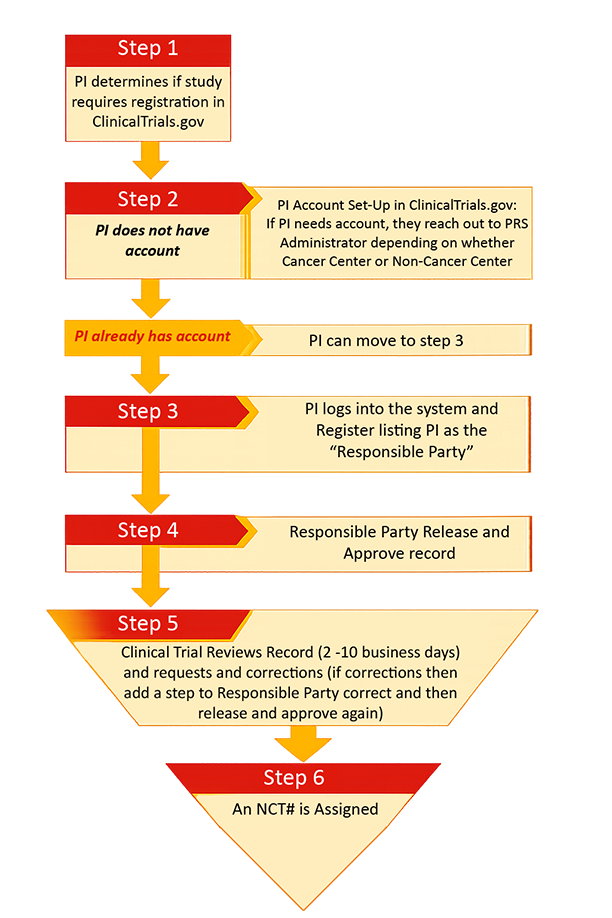
Consistency Checklist Process
DCG Review Process in I-STAR
For Non-Industry Clinical Trials & Agreements with Patient Care Costs Requiring an MCA
Why This Process Exists
- This process ensures that Informed Consent Form (ICF) language aligns with fully executed agreement terms for non-industry clinical trials involving patient care costs.
- It promotes consistency, compliance with sponsor and MCA requirements, protection of participants and the University, and proper documentation in I-STAR and Laserfiche.
- The review focuses on financial obligations, participant compensation, and subject injury coverage.
Roles & Responsibilities (Click here to download)
Stakeholder | Primary Role | Key Responsibilities | Point of Contact |
Study Team | Entering Information, | • Completes and enters accurate information in OnCore and I-STAR | |
Institutional Review | Human subjects | • Coordinates I-STAR completion with Study Team | Martin Koning-Bastiaan |
Department of Contracts | Agreement compliance | • Reviews, negotiates, and executes agreements | DCG Directory |
Clinical Trials Office | Medicare Coverage | • Creates and approves the MCA | Joanne Pak |
{kind=link}